|
Here is the infographics which will explain some detailed features on the HBB gene and where they are located, along with relations between the pre-mRNA and the final mRNA. 
Made using canva.com
0 Comments
The ship of Theseus: Ethics in Modifying Human Genome Recalling an interesting activity we did in genomics class two months ago, there were a few questions about my feelings towards editing my children’s genome sequence to make them stronger or smarter. I was instantly intrigued by the idea of modifying intelligence by altering genes sequence of an embryo. Do scientists simply insert a segment of genes that code for more neurons in the brain? However, a recent article helped me get closer to the answer. Genome-wide association studies(GWAS) is a study technique that allows scientists to look at vast amount of genetic variants and identify with variant is associated with specific traits. Since 2005 when the first GWAS study was successfully conducted, this technique has helped us identify certain locations of a lot of genetic disorders. Coming up with a specific section of genes that is responsible coding for intelligence is extremely difficult, and this trait called cognitive ability may spread in numbers of locations in the genome. Researchers used case-control setup to help more quickly identify the regions. They recruited over 100,000 people and screened their genomes, then compared the big data with over 300,000 more people in the database who had high-level achievements in intelligence. Cognitive ability, with relates to how smart the person will likely be, is an inheritable genetic code. For the first time, scientists can point out specific loci and say that those genes and influence the cognitive ability of a human. Also, some neuropsychiatric diseases are related to deletions and mutations in those certain loci of the genome, which prove the reliability of this GWAS study. With the advanced technology, we are even able to identify the locations of specific genes that are related to the intelligence level, which means we are one step closer to modifying them to make a super human. However, ethics concerns are being raised and heated up. Is it safe to alter those genes in human embryos? If so, wealthier people may have more chance to access this therapy, will genome editing define classes and social status of people by the quality of their modified genome? Like the ship of Theseus, are you still you if your component is already being tweaked? Source cited:
Newman, Tim. "'Cognitive Ability Genes' Identified". Medical News Today, 2017, https://www.medicalnewstoday.com/articles/320204.php?sr. "What Are The Ethical Concerns About Genome Editing?". National Human Genome Research Institute (NHGRI), 2017, https://www.genome.gov/27569225/what-are-the-ethical-concerns-about-genome-editing/. "Genome-Wide Association Studies Fact Sheet". National Human Genome Research Institute (NHGRI), 2015, https://www.genome.gov/20019523/genomewide-association-studies-fact-sheet/. “All models are wrong, some models are useful” We use candies to help us better model the process of the meiosis and mitosis, but there are some limitations in our video. The full process of cell duplication through mitosis and the sexual reproduction through meiosis and fertilization is extremely complicated. Our model is just a simplified version which we used one strand of candy worm to represent a complex structure of a sister chromatids. Two of the mistakes were from accidentally misspeaking two words. One of them occurred at 1:52, we meant to say “two centrosomes move to the opposite sides of the cell” instead of “two centromeres”. Then around 5:28, we said “two daughter cells are produced; however, these two daughter cells are not genetically identical to each other, which is different from meiosis”, it should be “which is different from mitosis”. These were due to misreading our script, and Allan went back and added the corrections in the description below the video. There are several more things I want to add besides the information provided in the video. Synthesis is important because for the cell to prepare for undergoing mitosis and meiosis, all the chromosomes have to be duplicate into pairs of sister chromatids. For organisms to go through sexual reproduction, cells have to go through meiosis and fertilization. However, we didn’t show the fertilization using our candies model, but we did do a voice-over explaining how meiosis is separating a parent cell into two gametes, while fertilization is when two gametes come together and form a zygote. Most of the citations for the pictures are mostly on the pictures. We did use some cake sprinkles to represent some other parts inside a cell. Since we only focused on the passing on of the DNA information, we zoomed in only on the DNA materials in the nucleus of the cell. Works Cited:
"Essentials Of Genetics Unit 2: How Does DNA Move From Cell To Cell?". Nature.Com, 2014, https://www.nature.com/scitable/ebooks/essentials-of-genetics-8/126042302#bookContentViewAreaDivID. Accessed 24 Feb 2018. Lab partner: Vaishnavi Kumar Introduction: PCR, short for polymerase chain reaction, is a useful way to help DNA scientists to amplify DNA segments and better understand it. Through PCR, we can make millions of copies of a specific region of DNA. So PCR is basically a special kind of DNA replication, but in a test tube!! Imagine if you have some hair or blood from a crime scene years ago, even the amount is minimal, as long as you can separate out a segment of DNA, then using PCR, you can “amplify” them and compare to match the criminals. In this lab, we will use enzyme Taq polymerase, which is very stable at high temperature. The results of the polymerase chain reaction are visualized using gel electrophoresis, the technique we practiced two weeks ago. Materials and Methods: Material list: 1% buffer TBE Gel tray Gel rig with comb 1 roll electrical tape Gloves Eppendorf tubes Micropipettor Thermocycler Enzyme “bead” Ice tank 10µL λ DNA Centrifuge Procedure Day 1 01.30.18 F Tue. Cast the gel and PCR We use 1% buffer TBE to cast the gel, and then carefully put them in a Ziploc bag and store in the fridge. Be sure to put on gloves for the PCR process, the oil on our hands can cause contamination which can affect the final results. We pair up into 3 groups of 2. For each big group, we have a control tube, which contains all the same contents, but unlike other test tubes, it doesn’t go through the thermocycler. We assign the well # to each pair. 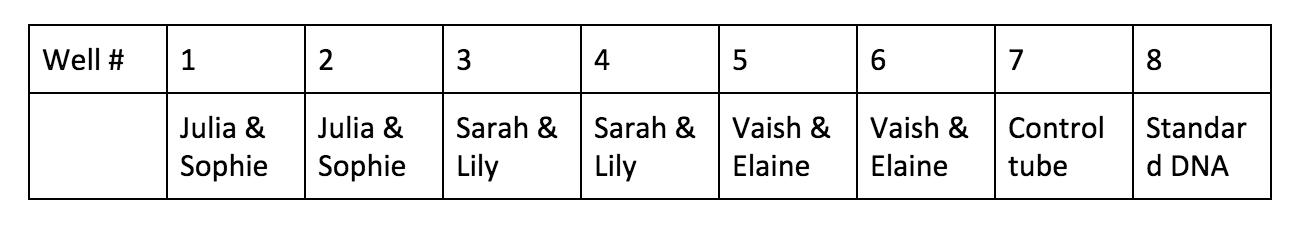 Obtain the 2 PCR tubes. We notice that there’s already substance inside the tube, which contains enzyme Taq polymerase, nucleotides and some salts needed for a successful reaction. Label each test tube, then put them into the ice holder.
Procedure Day 2 02.02.18 C Fri. Gel electrophoresis, visualize, and analyze the results: Add 4μL of UView loading dye to the PCR test tubes and the control tube. So after we finish the gel electrophoresis, we can be able to observe the DNA bands under the UV light.
Analyze the gel over UV light, here’s a photo of our results!  Results:  The distance traveled by each DNA bands is measured in centimeter. Be careful when measuring the standard DNA, which has has six fragments. * : an error occurred during the experiment.  The base pairs # of each of the six bands in the standard DNA is given by the teacher. Plot the point on a logarithmic scale graph.  Plot the curve according to the coordinates from known distance migrated and bas pairs # of the standard DNA. The line and dots in red means estimated values.  Predict/estimate the number of base pairs(y) using distance traveled(x) by extending the curve. * : an error occurred during the experiment. Conclusion: In this lab, we learned about how to perform PCR(polymerase chain reaction) using λ DNA and what happened in the tiny test tube and the control test tube. After this lab, I realize the whole experiment is mainly divided into two parts. First, the polymerase chain reaction with the help of thermocycler. Second, a visualized version of what the reaction looks like using the gel electrophoresis and UV dye helps to let our eyes better see and analyze them. For the reaction to successfully happen, it’s crucial for the test tubes to go through thermocycler because the DNA had to denature at the right temperature and the enzyme polymerase works the best under certain lower temperature. To prove this, we conduct a control tube(well #7) which doesn’t go through temperature changes in the thermocycler. From the results picture above, it’s clear that in all the successfully reactions(well #3-6), DNA bands traveled around 3.20-3.25cm. However, the mixture in control tube(well #7)doesn’t travel that far although it has all the contents needed for the reaction to happen. The control test tube(well #7) teaches us the importance of going through the right temperatures, and the error occurred with Julia and Sophie’s test tubes(well #1&2) also shows us the importance of the presence of BOTH the anneal primers and the λ DNA. According to Julia and Sophie, they misread the instructions and added only the anneal primers into their first test tube(well #1) and only the λ DNA into their second test tube(well #2). Though both test tubes had the Taq enzymes, nucleotides, and other chemicals required for a successful reaction, and went through the temperature cycles in the thermocycler, the results are not positive. The first test tube with only the anneal primer(well #1) appears on the final gel results but didn’t really move; the second test tube with only the λ DNA didn’t even appear on the final gel picture. To avoid this type of experimental error, be sure to read the instructions diligently and make sure to understand the importance of the presence of all the chemicals. Below are several questions that may help guide me for further investigation: What is crucial about Taq polymerase to the PCR? What does Taq polymerase make out of? Why makes it stable at high temperature? What does each part of “denature - anneal - extend” means? What do the Taq polymerase, the primer, and the λ DNA look like in each of the stages? What does it mean by “anneal”? What substance “extends”? Discussion: PCR, polymerase chain reaction, is the repetition of the cycle “denature - anneal - extend” over and over again in order to make millions of copies of DNA in short time and amplify a certain region of DNA so we can better analyze and understand it. The first step, denature, uses the special feature that DNA has at high temperature: it denatures and becomes single-stranded at around 95℃. Then the second step, anneal, the temperature is readjusted to where the primer will best react with DNA, which is around 55-60℃. Thirdly, extend, the Taq polymerase extends the primer at 5’-3’ direction and duplicates the DNA fragments between primers. The Taq polymerase will “synthesizes -- builds -- two new stranded DNA, using the original strands as templates”(NHGRI). Then each of these new strands can be used to create two new copies, and so on. The cycle of denaturing and synthesizing new DNA is repeated, sometimes “as many as 30 or 40 times, leading to more than one billion exact copies of the original DNA segment”(NHGRI). From the picture of gel, we can tell the significant difference between test tubes that were in the thermocycler(well #3-6 at around 3.20cm, 1540-1570 base pairs) and the control tube that wasn’t(well #7 at around 1.22cm, 23,130 base pairs). Without putting the control test tube into the thermocycler, the mixture in that tube did NOT go through any of the denature - anneal - extend cycle at all. Though inside the control tube has all the contents needed for the reaction, such as Taq polymerase, primers, and λ DNA, the chain reaction can’t be performed without going through the right temperatures for the “denature - anneal - extend” cycle.  Polymerase Chaine Reaction: Image credit: https://www.genome.gov/images/content/pcr_factsheet.jpg DNA replication process in the cell and same process in the test tubes in PCR share resemblances, but there are also differences. Firstly, to unzip the double strands into a single strand. In DNA replication in the cell, helicase unzips the double-stranded molecule. However, it’s costly to produce. In PCR, we use another solution to unzip the DNA: heat denature at around 95℃. Secondly, the RNA primer, a signal needed for DNA polymerase to start combining nucleotides onto the single strand inside an organism’s cell. In PCR, thousands of specially designed primers can anneal to the DNA when the temperature is cooling down from previous denaturing step. Thirdly, the polymerase. In the cell replication, DNA polymerase is responsible keep combining nucleotides and extend the growing DNA strand. In PCR, we still use DNA polymerase, but a special one called Taq DNA polymerase.  Thermus Aquaticus. Picture credit: https://en.wikipedia.org/wiki/Thermus_aquaticus#/media/File:Thermus_aquaticus.JPG Taq polymerase comes from the Thermus aquaticus, a species of bacteria that live in hot spring and boiling mud. Taq polymerase derived from these bacteria is a milestone discovering. Because like the bacteria in its name, Taq DNA polymerase is very thermostable even at the high temperature when DNA denatures. Other polymerase enzyme derived from some other bacterium would have already denatured during the process of unzipping DNA. It can also be used repeatedly around 25-30 times before it breaks down. Taq polymerase not only stay stable under high temperature, it also makes the PCR much more efficient and cost-effective. DNA is naturally colorless, so it must be stained so that we can better visualize the DNA bands and their locations. The UV dye we used makes the DNA bands glow under the UV light, which is much easier for us to do all the measuring and analysis. Some “Tracking dye which also contains a component(usually glycerol or sucrose) to increase the density of the sample to facilitate the loading”(bioinformatics). The known size of the amplicon is 1,106 base pairs given by our instructor, which is around 400 base pairs fewer than our closest results. There are a few possible inaccuracies which can lead to bigger experimental # of base pairs than the actual value. Since we plot the graph on a logarithmic scale by hands, and we have to estimate the extended graph, it’s very likely that the point deviates the best fit line. Extended BONUS question: Use the following information to calculate how many target DNA molecules were present in the 4.0 nanograms of λ DNA that you added to your PCR reaction:
  Works cited and reference: "Polymerase Chain Reaction (PCR) Fact Sheet." National Human Genome Research Institute (NHGRI), 2015, https://www.genome.gov/10000207/polymerase-chain-reaction-pcr-fact-sheet/#al-1.
"PCR." Sci.Sdsu.Edu, 2002, http://www.sci.sdsu.edu/~smaloy/MicrobialGenetics/topics/in-vitro-genetics/PCR.html. "Polymerase Chain Reaction (PCR)" Biology Animation Library :: DNA Learning Center." Dnalc.Org, https://www.dnalc.org/resources/animations/pcr.html. "Polymerase Chain Reaction (PCR)." Khan Academy, 2018, https://www.khanacademy.org/science/biology/biotech-dna-technology/dna-sequencing-pcr-electrophoresis/a/polymerase-chain-reaction-pcr. "Polymerase Chain Reaction (PCR)." Biology-Pages.Info, 2014, http://www.biology-pages.info/P/PCR.html. "Openstax CNX." Cnx.Org, 2016, https://cnx.org/contents/[email protected]:exg4e4AU@7/Biotechnology. "Polymerase Chain Reaction (PCR)." Ncbi.Nlm.Nih.Gov, 2017, https://www.ncbi.nlm.nih.gov/probe/docs/techpcr/. "Polymerase Chain Reaction / PCR | Learn Science At Scitable." Nature.Com, https://www.nature.com/scitable/definition/polymerase-chain-reaction-pcr-110. "The Role Of Taq Polymerase In PCR." Sciencing.Com, 2017, https://sciencing.com/role-taq-polymerase-pcr-7298417.html. Phillips, Theresa. "Visualizing DNA." The Balance, 2018, https://www.thebalance.com/visualizing-dna-375499. "Electrophoresis." Bioinformatics.Nl, http://www.bioinformatics.nl/molbi/SimpleCloningLab/electrophoresis.htm.
"Thermus Aquaticus." En.Wikipedia.Org, https://en.wikipedia.org/wiki/Thermus_aquaticus#/media/File:Thermus_aquaticus.JPG. By Allan Kalapura, Vaishnavi Kumar, Elaine Wang DNA, short for deoxyribonucleic acid is an extremely complex molecule that contains all the information necessary to build and maintain an organism. It serves to pass on genetic information from parent to offspring, a characteristic known as heredity. DNA is composed of smaller subunits called nucleotides, of which there are four varieties. Nucleotides are made up of three main components, a 5-carbon sugar molecule called deoxyribose, a phosphate group, and a nitrogenous base. The nitrogenous base is responsible for the identity of the nucleotide. DNA is composed of four types of nucleotides: adenine (A), cytosine (C), guanine (G), and thymine (T). These nucleotides bond in pairs, with A and T only bonding to each other, and G and C bonding to each other. In total, there are over 246 million nucleotides in one chromosome, and over 6 billion in one cell. The picture below shows the structure of DNA in detail. In order for DNA to be passed on from parent to offspring, it must be replicated.  The process of DNA replication starts with helicase, an enzyme that pulls apart the double stranded DNA. It starts in an area rich with A-T pairs since these are easier to separate because they contain two hydrogen bonds as opposed to C-G pairs that contain three hydrogen bonds. Once the double stranded structure is split, two DNA polymerase enzymes collaborate to copy the leading strand and the lagging strand. On the leading strand, DNA polymerase binds the nucleotides in 5’-3’ direction, while RNA primase inserts starter RNA primer at the initial point, giving the DNA polymerase a signal to start adding the corresponding nucleotides to the strand. On the lagging strand however, RNA starts the binding process. DNA polymerase has to work backwards in 3’-5’ direction, resulting in okazaki fragments, which are short DNA segments that make up the lagging strand of the newly synthesized DNA. After finishing one okazaki fragment, the “clamp” that secures DNA polymerase to the lagging strand dissociates and lets DNA polymerase release the lagging strand temporarily. As the double stranded structure keeps getting split by the helicase, the RNA primase is initiated and inserts a short RNA primer. Then DNA polymerase clamps back to the lagging strand again and begins working from where the primers are. The polymerase stops when it reaches the point where the preceding okazaki fragment is and releases the lagging strand, waiting for RNA primase to signal. Once the copying work is done, an enzyme called exonuclease takes away the RNA primer, and DNA polymerase replaces the gap with DNA nucleotides. At the end of the process, ligase fill the gaps left in the sugar-phosphate backbone.  The structure of DNA easily facilitates the function of replication. Despite the strong double helix that stabilizes the base pairs inside the structure, the weak hydrogen bonds that hold the base pairs together allow the molecule to untwist easily for replication. The DNA molecule is split down the middle into two strands and is now able to create two copies of DNA. Various enzymes are present to stimulate the reaction as the base pairs dislodge from each other. Since the nucleotides are exposed, corresponding base pairs match up to recreate the ladder structure. Once this is complete, this newly generated DNA strand coils back up into the double helix.  DNA replication is a crucial part of cell reproduction in all living organisms. In fact, life is dependent on this because without replicating DNA, our information would not pass down to future generations and life would no longer exist.
Sources:
Lab#1: Measurements, Micropipetting, and introductory Electrophoresis for DNA Labs Lab partners: Vaishnavi Kumar, Gray Martucci. Introduction: The purpose of this lab is to learn several important techniques performed in DNA labs such as using micropipettor to transport small amount of liquid, casting the gel, loading the wells accurately using micropipettor, and gel electrophoresis. Although we used candy dye instead of actual DNA in this lab, the basic concept of how the dye elements are separated by colors is the same as how DNA segments are separated by different sizes. Similarly, both the dye and the DNA samples are loaded into the wells, and “an electric current is applied to pull them through the gel. DNA fragments are negatively charged, so they move to the positive electrode. Because DNA fragments have the same amount of charge per mass, smaller fragments move through the gel faster than larger ones.”(Gel electrophoresis) This technique helps scientist visualize DNA fragments since they can see the groups of DNA fragments with the same size in bands. “Using electrophoresis, scientists can understand how many different DNA fragments are present in a sample and how large they are relative to one another.”(Gel electrophoresis) Source cited: "Khan Academy." Khan Academy, 2018, https://www.khanacademy.org/science/biology/biotech-dna-technology/dna-sequencing-pcr-electrophoresis/a/gel-electrophoresis. "What Is Gel Electrophoresis?." Yourgenome.Org, 2018, https://www.yourgenome.org/facts/what-is-gel-electrophoresis. Lab procedure: 01.16.18 B Tue. Preparing the gel, extracting the food dye from candies, and preparing four samples with standard color dye: We start with pouring the melted agarose gel into the gel tray wrapped with electrical tape, put the comb in it to cast 9 wells. When are waiting for the gel to solidify, we use dye extraction solution to get the food dye on the M&M candy coating. Once we get the dark solution, remove the candy from the cup. Pipette the solution into labeled microcentrifuge tube. Use a micropipettor size 2-20µL, we prepare four sample tubes A, B, C, and D each with different amount of four standard dyes, Blue 1, Yellow 5, Yellow 6(orange), and Red 40. Centrifuge all the test tubes and sit them on a tube rack. 01.17.18 C Wed. Loading the gels and Gel electrophoresis: First we take out the comb from the gel, put the solidified agarose gel with the tray into a gel rig, pour a 0.892 TAE buffer so all parts of the gel are immersed under the buffer. Use micropipettor size 2-20µL, load around 10-15µL of each sample carefully into each wells from left to the right(label different sides of the gel rig). Close the lid of the gel rig, plug it into power source and start electrophoresis. Starting time: 10:28am, with a voltage of 100V. After around 17 min, we observe that most of our samples have traveled obvious distance to the positive side, we turn off the power source and end the electrophoresis. We take the gel out of the gel tray and take a picture of the gel on the light screen. Results: Data table: (we print out the photograph and measure from paper.)   Conclusion:
In the four sample test tube, A, B, C, and D, blue color dye bands all appear which matches our prediction. They are also the closest to the wells, which shows that blue dye molecules have the biggest size among all. Below all the blue bands, well A, B, and D show orange bands, too, which matches our table since we add yellow 6(orange) dye to test tube A, B and D. Close to the orange dye, only well C shows an obvious red band while we did add red 40 dye to test tube A, C, and D. An educated guess will be in test tube A and D, yellow 6(orange) may travel to a very similar distance to the red 40 and our eyes can't pick up the color difference. All the dye molecules in our seven samples migrate towards the positive electrode, meaning the molecules are negative. The dye molecules which travel the fastest are the yellow dye molecules; the dye molecule travel the shortest are the blue molecules. The distance traveled varies by different type of the molecules. This is because shorter, smaller molecules travel through the agarose gel pores faster that long, bigger molecules do. In both my lab partner Gray and Vaishnavi’s candy samples, yellow 5 dye bands appear. The results matches our M&M candy colors we chose, too. Gray picks a green one, and blue and yellow bands show up; Vaishnavi picks a yellow candy, and only yellow band appears; I chose a orange candy, so only yellow 6(orange) dye shows up. Discusion: The electric current that runs through the buffer and the gel forces the charged molecules in the gel to move to opposite charged ends. In DNA molecules, the charge and the size influence how the dye molecules migrate in an agarose gel. Also, the agarose gels have the right size of dense pores that allows molecules to squeeze through if they are small enough, while bigger molecules are trapped and stay at the same place and form a “band” which can be visible to us. The charge changes the direction of the migration. Negatively charged dye molecules will be pulled towards the positive electrode, while some positively charged molecules will travel to the negative side. The size influence the distance traveled by the molecules, the smaller the molecules are, the faster they travel through the agarose gel pores, resulting in longer distance traveled, and vice versa. Among the carminic acid, betanin, fast green FCF, and Citrus Red 2 those four dyes, the Fast green FCF will be the most suitable for our lab. Since this dye has a negative charge, it will move towards the positive end of the gel and makes it more obvious for us to see. To prepare for our color dye gel electrophoresis experiment, we cast the wells in the middle of the gel. Different from color dyes molecules that may be negatively or positively charged, the DNA molecules are all negatively charged. When we are using DNA molecules to perform the gel electrophoresis, we only have to cast the wells at one end, and face it towards the positively charged electrodes in order for all DNA molecules to run towards the positive end. Dalton is a unit for atomic mass. DNA molecules with larger daltons like 5000da will move slower to the positive end of the gel, resulting in a short distance from the well. DNA molecules with a smaller daltons like 600da will move faster, resulting in a long distance far away from the wells. The electrophoresis allow the scientists to directly measure the daltons of DNA fragments by calculating their distance traveled, therefore, scientists can compare different sizes of DNA fragments. Bibliography: Works cited: "Khan Academy." Khan Academy, 2018, https://www.khanacademy.org/science/biology/biotech-dna-technology/dna-sequencing-pcr-electrophoresis/a/gel-electrophoresis. "What Is Gel Electrophoresis?." Yourgenome.Org, 2018, https://www.yourgenome.org/facts/what-is-gel-electrophoresis. "Unified Atomic Mass Unit." En.Wikipedia.Org, 2018, https://en.wikipedia.org/wiki/Unified_atomic_mass_unit. In the book Crack in Creation, there is an interesting story. When a variety of scientists were having a conference about the advantages and the dangers of presenting genome-typing technology in front of public, they got into a fierce debate about the morality of editing human genomes. While majority of the scholars was worried about the practicality and ethicality, one of the scientists boldly guessed that what if years later it’s immoral to not use this technology and edit the human genomes. Tie to the current world, this story reminds me that twenty years ago, people still thought that having an IVF(In Vitro Fertilization) baby was scary and immoral. However, it has gradually become accepted by people, especially couples that don't have the fortune to have babies themselves.
Personally, I would be comfortable having my gene tested for both ancestry and medical information and I would be very supportive of my future spouse having his or her genome sequenced. So far I don’t quite understand the purpose of having my full gene sequence besides saving my genetic data in my electronic medical records. I consider my gene data as my most unique ID, and it should be kept private only to myself. However, the genome-typing technology can bring potential moral problems when it is used to assist reproduction. Though I am comfortable with IVF, editing genome of my child or any child in their embryonic stage to give them an advantage, the idea of making a superhuman is the line that I can’t cross. Here are many questions I have considering the genome-editing technology, and this blog page will record things I've learned in the field of genomics and genome-typing. If editing the gene sequence of crops and non-human organisms in order to serve food for human and prevent disease is ok, then what are the differences of editing human genomes than editing non-human genomes? Both technically and morally? If we start to using genome-typing technology on embryonic stage human only to delete the flawed genes to prevent diseases, like malaria, what is the line between deleting faulty genes and editing other gene sequence to produce superhuman? Will anyone cross this line once he or she controls this technology? How to introduce this unprecedented genome-editing technology to the public and will they accept it? Will bringing the extinct species back using genome-editing technology be immoral? Will using the genome-typing technology become more cost-effective overtime? What is the future like with this life-changing technology? I believe my generation can witness the progression, or maybe the crisis? This blog will keep track of the evolution and along with my journey exploring in genome-typing technology. Work cited: DOUDNA, JENNIFER A. CRACK IN CREATION. [S.L.], MARINER BOOKS, 2018,. |
AuthorHello, I am Elaine. A junior at Holland Hall School. I am passionate about learning genomics and genome editing. In this blog page, you will see lab reports, personal opinion pieces, and some essays on biology! Archives
April 2018
Categories
|






















 RSS Feed
RSS Feed

{kind=link}